
Along the pathway from DNA to functional protein, much is already understood. DNA is transcribed into RNA which is then translated into the primary structure of a protein. Similarly, for many proteins the mechanisms by which they function is well documented. However, the processes by which an unfolded string of amino acids folds into a complex secondary and tertiary structure is still shrouded in mystery.
Unfortunately, the variable, dynamic, and short-lived nature of even the most important and stable states along the folding pathway make experimental analysis difficult. Even in simulations, the folding pathway cannot be easily examined because of the incredible computational demand of checking all possible steps that might be taken by the folding protein. These problems are addressed using the microscopically reversibility of protein folding. By studying unfolding pathways in molecular dynamics simulations—a much less computationally intensive task—a great amount of insight can be gained about the folding pathway.
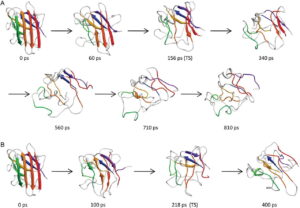
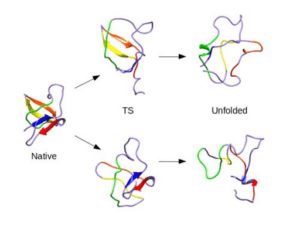
Towards this end, we have simulated over 800 distinct proteins under both native and unfolding conditions for a total of over 6000 simulations. For over 1300 of these simulations representing more than 180 distinct proteins, the transition state has been identified and characterized, and for five of these the transition state was verified against available experimental data. When general properties such as solvent exposed surface area and number of native contacts were calculated for all of these transition state ensembles, it was found that the standard deviations were small indicating that there are common rules influencing the structure and properties of all transition states and folding pathways. The overarching goal then becomes to elucidate these rules through the further study of molecular dynamics simulations.